Doyle Lab Creates Open-Source Software To Optimize Reactions
In the past few years, the Doyle Lab has turned increasingly to data science techniques to assist problem-solving in organic synthesis. Researchers are driven partly by a year-old federal initiative that seeks to conjoin data science and chemistry, and partly by the notion that a chemist’s time is better spent exploring new reactions than optimizing them.
Using that mission to help synthetic chemists at the bench, researchers have developed an open-source software tool that provides them with a state-of-the-art optimization algorithm for everyday work, folding what’s been learned in the machine learning optimization field into synthetic chemistry.
The software adapts key probabilistic principles of Bayesian Optimization (BO) to allow faster and more efficient syntheses of chemicals. BO is a sequential decision-making algorithm that balances the exploration of an experiment’s search space with the exploitation of information from available data.
In collaboration with the Adams Lab in Princeton’s Department of Computer Science and colleagues at Bristol-Myers Squibb, the work, “Bayesian Reaction Optimization as a Tool for Chemical Synthesis,” was published in Nature this week.

Photo by C. Todd Reichart
The paper includes a study comparing human decision-making capabilities with the software package. It found that the optimization tool yields both greater efficiency over human participants and less bias on a test reaction.
“Reaction optimization is ubiquitous in chemical synthesis, both in academia and across the chemical industry,” said Abigail Doyle, the A. Barton Hepburn Professor of Chemistry. “Since chemical space is so large, it is impossible for chemists to evaluate the entirety of a reaction space experimentally. We wanted to develop and assess BO as a tool for synthetic chemistry given its success for related optimization problems in the sciences.
“It’s all about using data to its fullest extent.”
Benjamin Shields, a former postdoctoral fellow in the Doyle lab and the paper’s lead author, created the Python package.
“I come from a synthetic chemistry background, so I definitely appreciate that synthetic chemists are pretty good at tackling these problems on their own. We tend to be less quantitative than other fields,” said Shields. “Where I think the real strength of Bayesian Optimization comes in is that it allows us to model these high-dimensional problems and capture trends that we may not see in the data ourselves, so it can process the data a lot better.
“And two, within a space, it will not be held back by the biases of a human chemist,” he added. “Where the human intuition kind of trickles into the problem is in that first step – you have to choose the space, you have to choose where you’re looking.”
How it Works
The software project started as an out-of-field proposal that Shields put together to fulfill doctoral requirements. Doyle and Shields then formed a team under the Center for Computer Assisted Synthesis (C-CAS), a National Science Foundation initiative launched at five universities to transform how the synthesis of complex organic molecules is planned and executed. Doyle has been a P.I. with C-CAS since 2019. The team received funding from the Princeton Catalysis Initiative to work on the idea.
“After initiating our collaboration with PCI and Professor Doyle, we learned of her efforts in C-CAS. This new NSF-sponsored center’s goals certainly aligned well with the interests we have at Bristol-Myers Squibb to leverage ML/AI techniques to better design robust, sustainable routes and speed drugs to patients,” said Jacob Janey, scientific director at BMS and an author on the paper.
Experimental, coding, and theoretical contributions by co-authors, including Ryan Adams, professor of computer science and P.I. of the Laboratory for Intelligent Probabilistic Systems, led to the successful development of the software.
“Reaction optimization can be an expensive and time-consuming process,” said Adams, who advised Shields on aspects of the research. “This approach not only accelerates it using state-of-the-art techniques, but also finds better solutions than humans would typically identify. I think this is just the beginning of what’s possible with Bayesian Optimization in this space.”
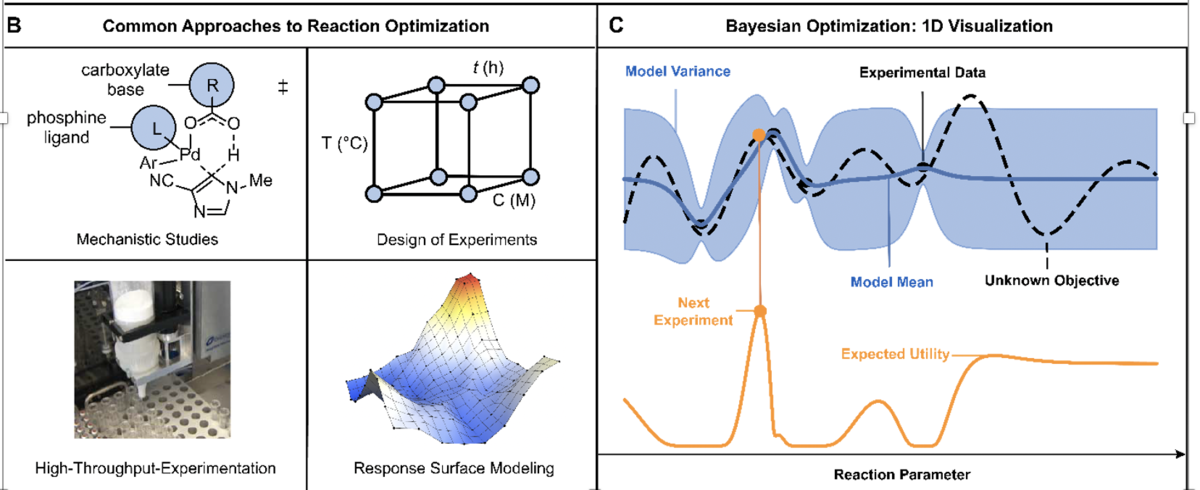
Figure credit: Nature
Users start by defining a search space, or, plausible experiments to consider, such as a list of catalysts, reagents, ligands, solvents, temperatures, and concentrations. Once that space is prepared and the user defines how many experiments to run at a given time, the software chooses initial experimental conditions to be evaluated. The user inputs the outcome of these experiments into the software, which then suggests new experiments to run, iterating through a smaller and smaller cast of choices until the reaction is optimized or there are no more gains to be realized.
Shields emphasized that as useful as the software may prove for synthetic chemists, it is still just a data tool – one that functions best when human expertise is guiding it.
“In designing the software, I tried to include ways for people to kind of inject what they know about a reaction,” he said. “No matter how you use this or machine learning in general, there’s always going to be a case where human expertise is valuable.”
The software and examples for its use can be accessed at this repository. GitHub links are available for the following: software that represents the chemicals under evaluation in a machine-readable format via density-functional theory; software for reaction optimization; and the game that collects chemists’ decision-making on optimization of the test reaction.
Read the full Nature paper here: https://www.nature.com/articles/s41586-021-03213-y.
“Bayesian Reaction Optimization as a Tool for Chemical Synthesis,” was authored by Abigail Doyle, Benjamin Shields, and Ryan Adams of Princeton University; Jason Stevens, Jun Li, and Jacob Janey of Bristol-Myers Squibb; Marvin Parasram of the University of Illinois at Chicago; and Farhan Damani of Johns Hopkins University.
This research was supported by funding from Bristol-Myers Squibb, the Princeton Catalysis Initiative, the National Science Foundation under the CCI Center for Computer Assisted Synthesis (CHE-1925607), and the DataX Program at Princeton University through support from the Schmidt Futures Foundation.