Hyster Lab Discovers Rare Mechanism to C-N Bond Formation Using Protein Engineering
Following is a summary of new research out in Nature from the Hyster Lab.
PAPER: “Emergence of a Distinct Mechanism of C-N bond formation in photoenyzmes”
JOURNAL: Nature, October 2024.
AUTHORS: Felix Raps, Ariadna Rivas-Souchet, Chey Jones, Todd Hyster.
WHAT IT IS:
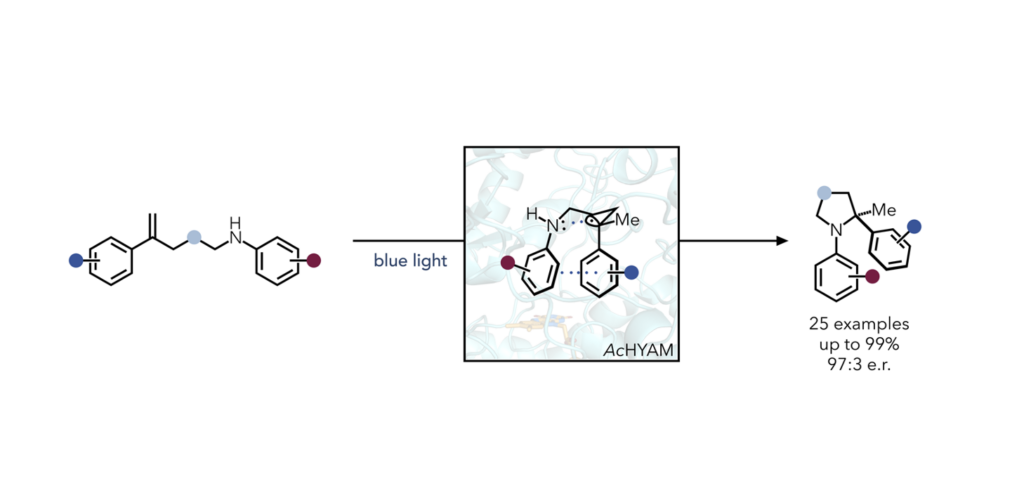
Continuing in their mission to find ways to engineer enzymes for non-natural transformations, researchers in the Hyster Lab found a “truly unexpected” emergent mechanism for the formation of C-N bonds, catalyzed by Baeyer-Villiger Monooxygenase. The mechanism is hypothesized to be available to a protein because of its ability to pre-organize a substrate.
Researchers are not aware of any such transformation in small molecule catalysis that uses this mechanism of C–N bond formation. Furthermore, chiral amines are found in up to 50% of small molecule drugs, so new methods to access these motifs have the potential to be extremely impactful.
HOW THEY DID IT:
This research highlights the capacity for enzymes to reveal new pathways to reactivity challenges in chemical synthesis. Researchers used a strategy of directed evolution to improve the proteins’ ability to perform this reaction by targeting residues within the active site for mutation. After five rounds of mutations, they achieved a variant that provided excellent product yield and stereoselectivity.
The research exploits a through-space interaction of a reductively generated benzylic radical and the nitrogen lone pair.
COMMENT FROM P.I. TODD HYSTER:
“In truth, this is probably the last mechanism that we thought it would be; we sort of went down the list of all the other reasonable mechanisms. This was truly unexpected. We found that C–N bond formation can occur through a carbon-centered radical intermediate. The way you do this in solution would be to oxidize that radical to a cation, which is trapped by the amine.

Professor of Chemistry Todd Hyster.
“But we have fairly good evidence suggesting that’s not what’s going on in this case. Instead, the protein is using a series of non-covalent interactions to pre-organize the substrate, enabling the radical to interact with the nitrogen. And it turns out that this interaction makes the radical easier to oxidize, resulting in C–N bond formation.
“This transformation doesn’t involve a pre-activated substrate. We haven’t introduced functionality that we’re going to throw away during the reaction. If I were to show this to a synthetic organic chemist, most would say, ‘You’re oxidizing the alkene or the amine to achieve C-N bond formation.’ But in this case, it’s the other way around: we’re actually reducing the alkene and that is what’s driving the reaction forward.”
COMMENT FROM FELIX RAPS, POSTDOC AND LEAD AUTHOR ON THE PAPER:
“We tried to translate activity that pioneers in small molecule catalysis have discovered, including Robert Knowles’ work on hydroaminations. Our goal was to get an enzyme to perform this reaction to address selectivity challenges associated with them. What we found was surprising in that we did not see the product we would expect from this precedent.

Postdoc and lead author on the paper Felix Raps.
“Our enzyme catalysts consist of amino acid units that we can modify to change the structure and activity. This process of directed evolution allows us to make our enzymes better at this activity by iteratively modifying single sites. We then explored the mechanism with this optimized enzyme by designing modified substrates which can give us evidence in ruling out certain mechanistic possibilities. We ended up with a full picture using simulations with our great collaborators from Merck.”
COMMENT FROM CO-AUTHOR ARIADNA RIVAS-SOUCHET:
“For me, the main takeaway of this project is how enzymes can enable new mechanisms that are otherwise not accessible to small molecule catalysis.
“I personally worked on developing the scope as well as the mechanistic studies. It was very exciting to dive into the mechanistic studies and uncover something unexpected.”
DOI FOR THE PAPER IN NATURE: https://doi.org/10.1038/s41586-024-08138-w
FUNDING: This work was supported by the National Institutes of Health (NIGMS R01GM127703).