Knowles Converts Stable Olefins to Less Stable, Solving Standing Challenge
Chemists have long sought methods to convert more stable internal olefins into less stable terminal olefins. Isomerization reactions that proceed against a thermodynamic bias, as this one would, are generally challenging for conventional thermal catalysis.
With an innovative use of photocatalysis and chromium co-catalysis to provide an energy offset, however, the Knowles Lab reports a general method to generate less-stable olefins from internal olefins catalytically.
Researchers used excited-state electron transfer events to “pump” olefin molecules up and then have them fall back down through a series of favorable steps to ultimately transform into a less stable isomeric form. This essentially allows internal olefins, or alkenes, to migrate along the carbon chain to the terminal position—a kind of musical chairs that shunts the double bond to a position of less overall stability.

Kuo Zhao, a fifth-year graduate student in the Knowles Lab and lead author on the paper.
The research marks an exciting development because terminal olefins are useful starting points in a wide range of chemical processes and as building blocks in industrial products. This research gives scientists a new tool to generate more of them.
The lab’s paper on the research, “Contra-Thermodynamic Positional Isomerization of Olefins,” was recently published in the Journal of the American Chemical Society (JACS).
The realization of contra-thermodynamic alkene isomerization has been a long-standing challenge for catalysis. By combing through and then combining some of the precedents he found in literature, Kuo Zhao, a fifth-year graduate student in the lab, was able to come up with a light-driven method for the direct ‘uphill’ isomerization of numerous olefin classes.
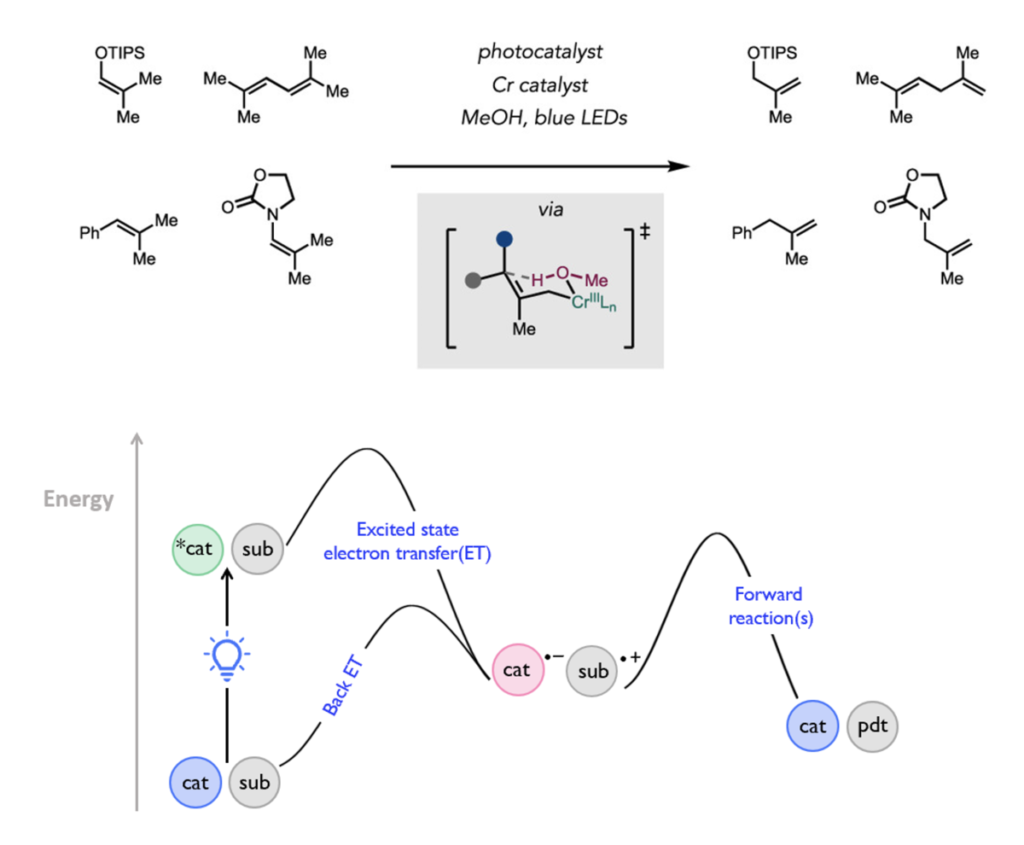
This image shows the contra-thermodynamic positional isomerization of olefins enabled by excited-state redox catalysis and chromium catalysis.
In the paper, Zhao describes how stepwise proton coupled electron transfer (PCET) activation of a more thermodynamically stable olefin substrate is mediated by an excited-state oxidant and a Brønsted base to give an allylic radical. That radical, in turn, is captured by a chromium (II) co-catalyst to generate an allylchromium(III) intermediate, which eventually undergoes regioselective protodemetalation to deliver a less-stable terminal alkene.
“This work is another example from our lab of using photochemistry to accomplish an uphill process. Traditionally, this is impossible, by definition, using conventional ground state catalysis. But with photochemistry, this can be achieved catalytically,” said Zhao. “When I looked back into it, I found from the literature that there’s a way to solve this problem by introducing chromium catalysis.
“Basically, you generate an allyl chromium intermediate and do an in situ protodemetalation to get the terminal olefin, which is the olefin you want in this transformation.
“As to how to use it, we’ll leave that to future chemists,” Zhao added. “But we’re showing in this research that this previously impossible transformation is now possible.”
Zhao said he worked on the problem for about a year, dipping into literature and then trying a series of transition metal catalysts to drive the transformation, which resulted in a low yield of the desired olefin and sent him back to the drawing board. Other graduate students within the Knowles lab had been working on this problem over the years, utilizing only photoredox catalysis.
“The Knowles lab had been looking to solve this problem for a number of years, but with little success,” said Professor of Chemistry Robert Knowles. “By merging chromium catalysis and photoredox catalysis, Zhao was able to solve this challenging problem.”
Click here to read the full paper: https://pubs.acs.org/doi/10.1021/jacs.1c11681
“Contra-Thermodynamic Positional Isomerization of Olefins” was authored by Kuo Zhao and Robert Knowles, and supported as part of BioLEC, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Basic Energy Sciences (under Award #DE-SC0019370).