Method for Nucleophilic Fluorination Advanced by Doyle Group
The Doyle Group published research this week announcing a new twist on a classical method to install fluorine. The research advances the long-standing pursuit of mild methods for the late-stage introduction of nucleophilic fluorine into molecules, particularly for translation in radiofluorination chemistry.
Fluorine is a critical element in pharmaceuticals, agrochemicals, and materials because of the unique chemical properties it confers on organic molecules. It is often incorporated due to the exceptionally strong bond with carbon that increases molecular stability and allows scientists to manipulate the material and physiochemical properties of the molecule.
To facilitate the introduction of fluorine into a molecule of choice, synthetic chemists must often use costly electrophilic variants that can be reactive with biologically functional groups. In addition, fluoride is a recalcitrant nucleophile and a strong base, and therefore has historically limited access to a range of desirable structures.

Photo by C. Todd Reichart
The method advanced by the Doyle Group overcomes these limitations by using photoredox catalysis to generate a highly reactive intermediate that rapidly traps even poorly nucleophilic fluoride under “incredibly” mild conditions, said Abigail Doyle, the A. Barton Hepburn Professor of Chemistry.
The method provides access to a broad range of aliphatic fluorides, a common functional group in many pharmaceutical candidates, and could be readily adapted to radiofluorination techniques. Radiochemists work with radioactive molecules to study the location and metabolism of pharmaceuticals.

Photo by C. Todd Reichart
“The goal was to expand the set of reactions that radiochemists can use to access different carbon-fluorine motifs,” said Eric Webb, a fifth-year graduate student in the Doyle Group and the paper’s lead author. “All too often, there is a barrier to using new radiochemistry technologies. Radiochemists want the chemistry to ‘just work’. We wanted to devise a robust method so that so radiochemical facilities can quickly implement this technology and apply it across a range of diverse molecules.
“Synthetically, this method gives you access to alkyl fluorides that would be difficult to obtain with the other nucleophilic techniques.”
The paper appears in the Journal of the American Chemical Society (JACS). The research is the product of a collaboration between the Doyle Group and the PET Radiochemical Synthesis group at Bristol-Myers Squibb Research and Development in Princeton, NJ.
The method relies on N-hydroxyphthalimide esters as substrates. Past research has underscored the use of these molecules as precursors to reactive alkyl radicals. Upon single-electron reduction of the substrate by the excited state of the photoredox catalyst, the resulting radical can be reintercepted by the photocatalyst and oxidized to form a positively charged carbon that rapidly traps even weakly reactive fluoride.
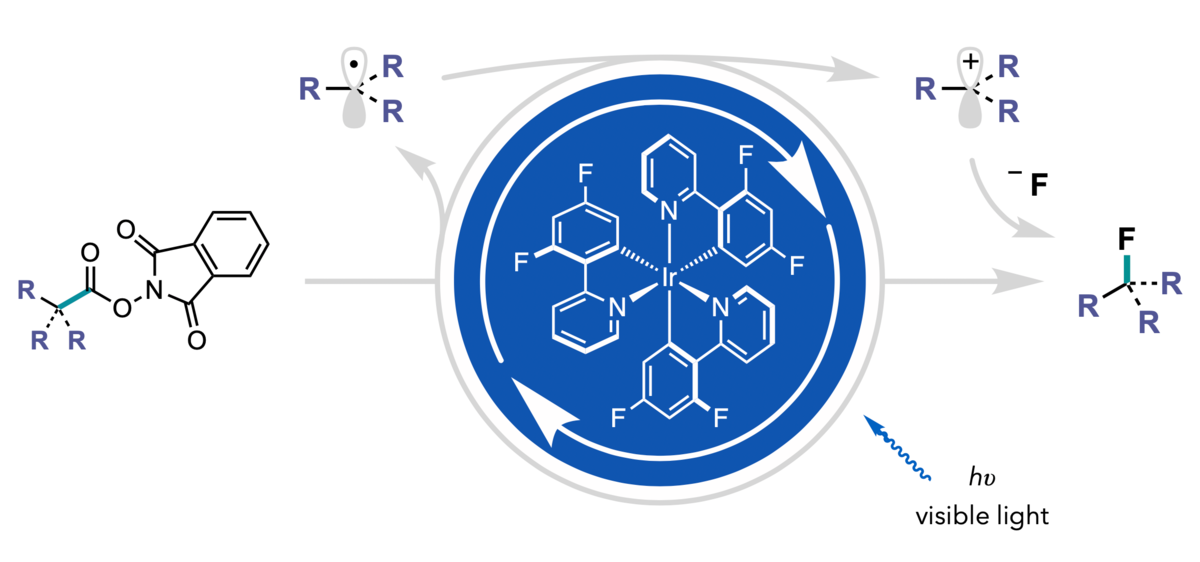
Illustration by Eric Webb
“Once the photocatalyst generates the radical intermediate, it really wants to take back an electron,” said Webb. “Since the photocatalyst can’t oxidize fluoride, the photocatalyst oxidizes the radical, and the negatively charged fluoride is attracted to the positively charged carbon, and readily forms a carbon-fluorine bond.”
This redox-neutral method for nucleophilic fluorination delivers primary, secondary, and tertiary benzylic fluorides, unactivated tertiary fluorides, and even some multifluorinated motifs with broad functional group tolerance.
The paper also describes mechanistic experiments that furnish evidence for both radical and carbocation intermediates and provides a strategy for other bond-forming methods.
The paper, “Nucleophilic (Radio)Fluorination of Redox-Active Esters via Radical-Polar Crossover Enabled by Photoredox Catalysis,” was authored by E.W., J.P., E.C., D.D., S.B., W.E., and A.D. Financial support was provided by NSF (CHE- 1565983) and Bristol-Myers Squibb. It can be read in full here: https://pubs.acs.org/doi/10.1021/jacs.0c03125