Putting two and two together
Researchers at Princeton have developed a cobalt-catalyzed [2π+2π] reaction that may give unprecedented access to cyclobutanes, four-membered ring-containing molecules. Previous [2π+2π] reactions, so named for their carbon-carbon double bond starting materials called alkenes, have been limited in scope, leaving many cyclobutane compounds out of reach, along with any potentially beneficial properties.
Led by Paul Chirik, the Edwards S. Sanford Professor of Chemistry, the team published the cobalt-catalyzed [2π+2π] reaction and a thorough investigation of its mechanism in the Journal of the American Chemical Society on June 1.
“Because examples of this reaction are so rare, we wanted to understand why these cobalt complexes were special and how they worked in the reaction,” said Valerie Schmidt, lead author on the article and a postdoctoral researcher in the Chirik lab.
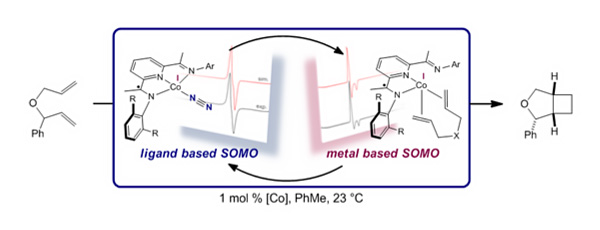
The new cobalt-catalyzed reaction overcame limitations that have plagued other transition metal catalyzed methods, such as poor selectivity or requiring very reactive alkenes as starting materials. The research team suspected their success came from the redox active bis(imino)pyridine ligands attached to cobalt, which are capable of passing electrons to and from the metal.
The Chirik group has used these redox active ligands previously, attached instead to iron to catalyze a [2π+2π] reaction reported in 2006. But the iron catalyst is highly sensitive to air and moisture, an issue that could be mitigated by switching to a less reactive metal like cobalt.
Replacing iron for cobalt presented a unique challenge in analysis because it altered the complex’s overall magnetic state from diamagnetic to paramagnetic. Unlike diamagnetic compounds, paramagnetic compounds can be difficult to identify by nuclear magnetic resonance (NMR) spectroscopy, a technique that uses a strong magnet to pulse atomic nuclei to reveal their environments, and a primary tool for characterizing molecules.
“We really had to be creative in finding ways to confirm our hypotheses about the catalyst,” Schmidt said. One extremely useful tool, analogous to nuclear magnetic resonance but pulses electrons instead of nuclei, was electron paramagnetic resonance (EPR). This technique allowed the researchers to track the unpaired electrons, called radicals, throughout the reaction.
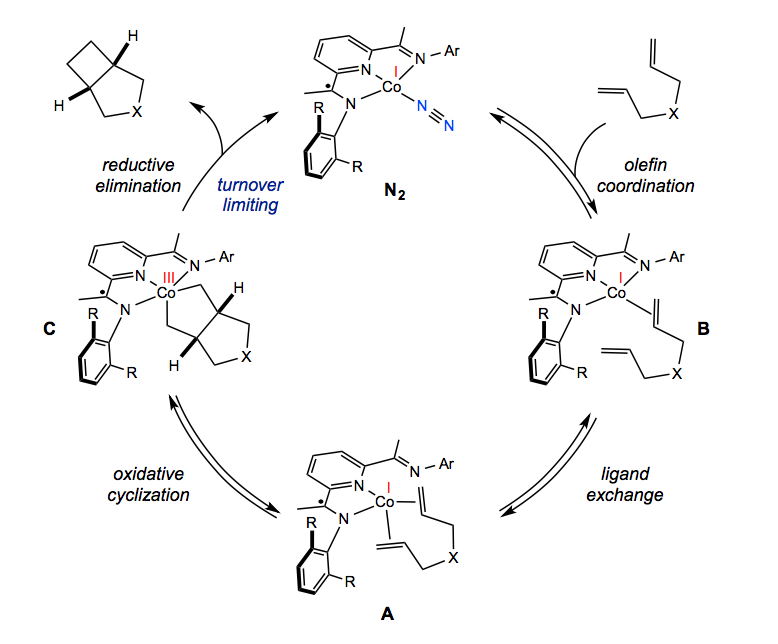
Additional data gathered from theoretical calculations to kinetic studies to x-ray crystal structure elucidation allowed the research team to sketch out a detailed reaction mechanism. They proposed that the cycle begins with successive coordination of the two tethered alkenes to the metal center.
Coordination of the second alkene was crucial, Schmidt explained, because it changed cobalt’s geometry, from square planar to tetrahedral, and effectively moved the unpaired electron from the ligand to the metal. Only then can the metal based radical promote the carbon-carbon forming event and push the reaction forward.
This action leads to the formation of a metallacycle—a pentagon shaped ring of four carbons and one cobalt atom. Cobalt is then squeezed out of the ring to release the final four-membered cyclobutane product in a process called reductive elimination. After testing a series of catalysts with varying size and electronic properties, the researchers suggested that reductive elimination was the turnover-limiting step, essentially the bottleneck of the reaction.
Armed with a deeper understanding of the cobalt catalyst system, the researchers hope to continually enhance its performance. “We want to make it as easy as possible to access cyclobutane containing molecules. Without this ability, we really have no idea what we are missing out on,” Schmidt said.
Read the full article here:
Schmidt, V. A.; Hoyt, J. M.; Margulieux, G. W.; Chirik, P. J. “Cobalt-Catalyzed [2π+2π] Cycloadditions of Alkenes: Scope, Mechanism and Elucidation of Electronic Structure of Catalytic Intermediates.” Journal of the American Chemical Society 2015, Article ASAP.
This work was supported by the National Institutes of Health Ruth L. Kirschstein National Research Service Award (F32 GM109594) and Princeton University Intellectual Property Accelerator Fund.